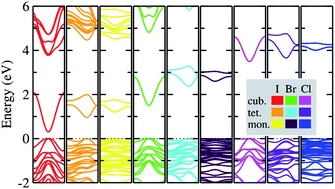
The vacancy-ordered double perovskites K2SnX6 (X = I, Br, Cl) attract significant research interest due to their potential applications as light absorbing materials in perovskite solar cells. However, deeper insight into their material properties at the atomic scale is currently lacking. Here we present a systematic investigation of the structural, electronic, and optical properties and phase stabilities of the cubic, tetragonal, and monoclinic phases based on density functional theory calculations. Quantitatively reliable predictions of lattice constants, band gaps, effective masses of charge carriers, and exciton binding energies are provided and compared with the available experimental data, revealing the tendency of the band gap and exciton binding energy to increase on lowering the crystallographic symmetry from cubic to monoclinic and on moving from I to Cl. We highlight that cubic K2SnBr6 and monoclinic K2SnI6 are suitable for applications as light absorbers for solar cell devices due to their appropriate band gaps of 1.65 and 1.16 eV and low exciton binding energies of 59.4 and 15.3 meV, respectively. The constant-volume Helmholtz free energies are determined through phonon calculations, which predict phase transition temperatures of 449, 433 and 281 K for cubic–tetragonal and 345, 301 and 210 K for tetragonal–monoclinic transitions for X = I, Br and Cl, respectively. Our calculations provide an understanding of the material properties of the vacancy-ordered double perovskites K2SnX6, which could help in devising a low-cost and high performance perovskite solar cell.