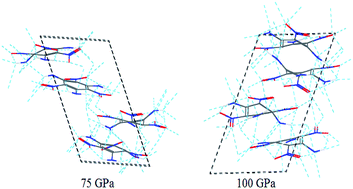
The crystal, molecular, and electronic structures of crystalline 1,3,5-triamino-2,4,6-trinitrobenzene (TATB) in the pressure range of 0–100 GPa under room temperature have been studied by ab initio molecular dynamics (MD). The DFT- and DFT-D-based MD were used to investigate the effects of the vdW correction on the results. The lattice parameters and P–V isotherm of TATB under compression by the DFT-D agree well with the experimental results, whereas the DFT without the vdW correction either underestimates or overestimates the results evidently. The DFT-D results show that TATB is chemically stable in the entire investigated pressure range, in agreement with the experiments, but the DFT without the vdW correction misestimates that TATB decomposes at 50 GPa and polymerizes at 100 GPa. Both the intra- and intermolecular hydrogen bonding are strengthened with the increasing pressure from 5 to 50 GPa, consistent with the experimental results. The DFT without the vdW correction misestimates that TATB turns into a metal system at 100 GPa. The PBE0 with the vdW correction was used to study the band structures of TATB, and the obtained results up to 50 GPa are in agreement with the available experiment results.