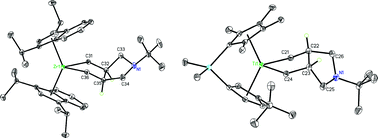
Metallation of a variety of α,ω-dienes has been explored with an η9,η5-bis(indenyl)zirconium sandwich compound and an ansa-titanocene dinitrogen complex. The η9,η5-bis(indenyl)zirconium sandwich compound, (η9-C9H5-1,3-Pr2)(η5-C9H5-1,3-iPr2)Zr, served as an isolable source of Negishi's reagent and readily formed a kinetic mixture of cis and trans diastereomers of the corresponding zirconacyclopentanes upon diene metallation. For pure hydrocarbon substrates such as 1,6-heptadiene and 1,7-octadiene, an equimolar amount of cis and trans diastereomers were the kinetic products; isomerization to the thermodynamically favoured trans isomers was observed over time at ambient temperature or upon heating to 105 °C, respectively. By contrast, substitution of the methylene or ethylene spacer in the α, ω-diene with a fluorenyl group (e.g. 9,9-diallylfluorene) resulted in exclusive kinetic formation of the trans diastereomer. Amino-substituted dienes were also readily cyclised and one example was characterised by single-crystal X-ray diffraction. Similar studies were also conducted with the ansa-titanocene dinitrogen complex, [Me2Si(η5-C5Me4)(η5-C5H3-3-tBu)Ti]2(μ2,η1,η1-N2), and both kinetic and thermodynamic selectivities evaluated. The use of a C1 symmetric ansa-metallocene increases the number of isomeric possibilities. For diallyl tert-butyl amine, diene metallation was more selective than for the bis(indenyl)zirconium sandwich compound and isomerization was also more rapid. Preliminary functionalisation reactivity for both the zircona- and titanocycles was also explored.