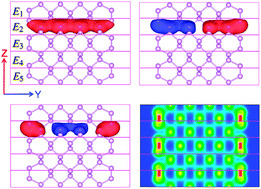
With the help of our recently developed massively parallel DGDFT (Discontinuous Galerkin Density Functional Theory) methodology, we perform large-scale Kohn–Sham density functional theory calculations on phosphorene nanoribbons with armchair edges (ACPNRs) containing a few thousands to ten thousand atoms. The use of DGDFT allows us to systematically achieve a conventional plane wave basis set type of accuracy, but with a much smaller number (about 15) of adaptive local basis (ALB) functions per atom for this system. The relatively small number of degrees of freedom required to represent the Kohn–Sham Hamiltonian, together with the use of the pole expansion the selected inversion (PEXSI) technique that circumvents the need to diagonalize the Hamiltonian, results in a highly efficient and scalable computational scheme for analyzing the electronic structures of ACPNRs as well as their dynamics. The total wall clock time for calculating the electronic structures of large-scale ACPNRs containing 1080–10 800 atoms is only 10–25 s per self-consistent field (SCF) iteration, with accuracy fully comparable to that obtained from conventional planewave DFT calculations. For the ACPNR system, we observe that the DGDFT methodology can scale to 5000–50 000 processors. We use DGDFT based ab initio molecular dynamics (AIMD) calculations to study the thermodynamic stability of ACPNRs. Our calculations reveal that a 2 × 1 edge reconstruction appears in ACPNRs at room temperature.