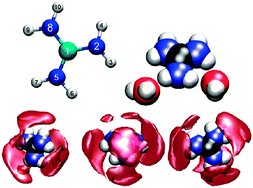
Structure and dynamics of guanidinium in aqueous solution were examined via a double zeta HF level Quantum Mechanical Charge Field-Molecular Dynamics (QMCF-MD) simulation, as well as two Molecular Mechanics-Molecular Dynamics (MM-MD) simulations, parametrised via the Amber99 parameter set, employing the side chain of arginine as a template. Coulombic parameters were fitted via Mulliken population data of the QM simulation, as well as via the recommended restrained electrostatic potential fit (RESP). Although guanidinium is one of the most weakly hydrated cations yet characterised, its hydration pattern is quite complex and pronounced in the quantum mechanical simulation. The positive charge is mainly located on the central carbon, resulting in strong solute–oxygen coordination. Hydrogen bonds are mainly donated by the amide hydrogens, but are also accepted via the nitrogens to a very low extent. Detailed analysis of structure and dynamics, comparing the applied QM and MM models, provides evidence that the arginine parametrisation leads to highly different results than the quantum mechanical treatment, and that the RESP parametrisation is too polarised.