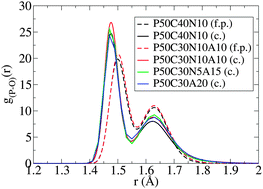
First-principles and classical molecular dynamics simulations of undoped and silver-doped phosphate-based glasses with 50 mol% P2O5, 0–20 mol% Ag2O, and varying amounts of Na2O and CaO have been carried out. Ag occupies a distorted local coordination with a mean Ag–O bond length of 2.5 Å and an ill-defined first coordination shell. This environment is shown to be distorted octahedral/trigonal bipyramidal. Ag–O coordination numbers of 5.42 and 5.54–5.71 are calculated for first-principles and classical methodologies respectively. A disproportionation in the medium-range phosphorus Qn distribution is explicitly displayed upon silver-doping via CaO substitution, approximating 2Q2 → Q1 + Q3, but not on silver-doping via Na2O substitution. An accompanying increase in FWHM of the phosphorus to bridging oxygen partial pair-correlation function is strong evidence for a bulk structural mechanism associated with decreased dissolution rates with increased silver content. Experimentally, Ag2O ↔ Na2O substitution is known to decrease dissolution and we show this to be a result of Ag's local bonding.