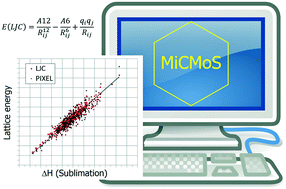
A set of 377 crystal structures extracted from the Cambridge Structural Database and matched to the experimental sublimation enthalpies of the corresponding materials is used to calibrate a new intermolecular potential field for organic compounds. The scheme includes usual R−6–R−12 Lennard-Jones dispersion–repulsion terms, with parameters for C, H, N, O, S and the halogens, fully transferable and unique for each atomic species without distinction among the hybridization states of carbon, or among valence states like carbonyl or ether oxygen or aza, nitro or nitrile nitrogen. This great economy in the number of parameters is countered by the need for using accurate coulombic terms over atomic point charges derived for each molecule from the fitting of the electrostatic potential (ESP method) of an ab initio MP2/6-31G** wavefunction. The potential scheme so derived, called Mi-LJC (Milano Lennard-Jones Coulomb), is able to reproduce the 377 sublimation enthalpies by the calculated lattice energies with an average percent deviation of 5% and a rmsd of 6.7 kJ mol−1. The potential scheme is also tested and proved applicable for the Monte Carlo simulation of some organic liquids, as well as for the molecular dynamic simulation of liquids and crystals.