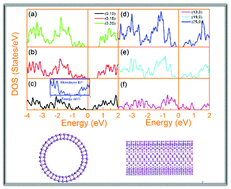
Due to its high carrier mobility and tunable bandgap, phosphorene has been the subject of immense interest recently. Herein, we show using density functional theory based calculations that black phosphorus (BP) nanotubes are achievable. Moreover, the electronic properties of BP nanotubes are explored. In contrast to their monolayer and bulk counterparts, most BP nanotubes possess indirect band gaps. In addition, strong anisotropic electronic behaviors are observed between zigzag and armchair nanotubes. Semiconducting to semi-metallic transition occurs only for zigzag tubes when its diameter shrinks to ∼1.5 nm. This difference is strongly related to the bond bending after the formation of the nanotubes which governs the s–p hybridization, as well as electron distribution in different p orbitals and this eventually determines the electronic structure of BP nanotubes.