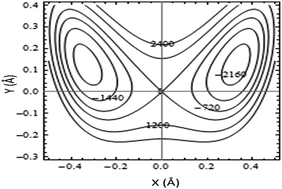
A two-dimensional potential energy surface was utilized to treat the proton transfer in acetylacetone (AA) and its α-halo derivatives: α-fluoro-(FAA), α-chloro-(ClAA), and α-bromo-acetylacetone (BrAA). This potential energy function, which couples O–H stretching and in-plane bending vibrations, was acquired through ab initio calculations for a fixed skeleton geometry. The resulting potential energy surfaces were then used to calculate the proton tunneling frequencies and proton transfer barrier heights. The barrier heights (the energy difference between the saddle point and the minima) calculated at the MP2/6-31G(2d,p) level of theory for proton transfers in AA, FAA, ClAA, and BrAA are 7.2, 9.4, 6.3, and 5.9 kcal mol−1, respectively. The theoretically predicted proton transfer barrier heights exhibit excellent linear correlations with geometrical, electronic structural, and topological parameters evaluated by the atoms-in-molecule (AIM) and natural bond orbital (NBO) analyses.