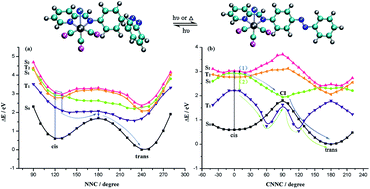
In this paper, we performed density-functional theory (DFT) at the B3LYP/6-311++G(d,p) & LANL2DZ quantum level to probe the thermal cis → trans isomerization of Re(CO)3–AB, a diimine ligand with an azo group substituent. We have searched for the transition states (TS) on the S0 and T1 potential energy surfaces and detected two inversion transition states TS-inv-1 and TS-inv-2 following a rotation-assisted inversion mechanism. In the T1 state, only the rotation pathway exists with the energy barrier 1.88 eV. We also investigated the photoisomerization pathway, the potential energy profiles of the vertical excitation for the excited states T1, S1, T2 and S2 were calculated, and we analyzed two possible isomerization routes through the inversion and rotation pathways. In addition, we probed the properties of the isomerization of the azo portion caused by excitation of the MLCT bands.