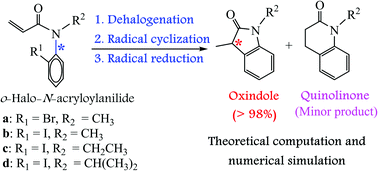
In this study, we employed density functional theory and numerical simulation methods to investigate the mechanism of intramolecular radical cyclization reactions of ortho-halo-N-acryloylanilides bearing N-alkyl substituents, namely, methyl, ethyl, and isopropyl. We established kinetic differential equations for elementary reaction pathways on the constructed potential energy profiles and performed numerical simulation computations. Using theoretically computed kinetic and thermodynamic data and numerically simulated regio- and stereochemistry, we revealed the detailed mechanism and memory of chirality of the investigated cyclization reactions. Furthermore, the levels of chirality transfer of ortho-halo-N-acryloylanilides with N-alkyl substituents larger in volume than isopropyl were numerically calculated. The computed results provided insight into the construction of nitrogen-containing heterocyclic compounds via the intramolecular radical cyclization approach using N-alkyl-substituted ortho-halo-N-acryloylanilide derivatives as the substrates.