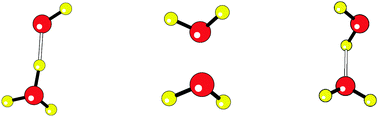
Due to the severe self-interaction errors associated with some density functional approximations, conventional density functionals often fail to dissociate the hemibonded structure of the water dimer radical cation (H2O)2+ into the correct fragments: H2O and H2O+. Consequently, the binding energy of the hemibonded structure (H2O)2+ is not well-defined. For a comprehensive comparison of different functionals for this system, we propose three criteria: (i) the binding energies, (ii) the relative energies between the conformers of the water dimer radical cation, and (iii) the dissociation curves predicted by different functionals. The long-range corrected (LC) double-hybrid functional, ωB97X-2(LP) [J.-D. Chai and M. Head-Gordon, J. Chem. Phys., 2009, 131, 174105], is shown to perform reasonably well based on these three criteria. Reasons that LC hybrid functionals generally work better than conventional density functionals for hemibonded systems are also explained in this work.