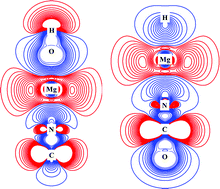
In this study, the structures and stabilities of [H,Mg,N,C,O] isomers were theoretically characterized by density functional theory and coupled-cluster computations. Linear magnesium isocyanide hydroxide (HOMgNC, 1) and magnesium isocyanate hydride (HMgNCO, 4) molecules in electronic singlet states exhibit considerably high kinetic stabilities. Electronic structure analyses clearly confirmed the ionic natures of the Mg–X bonds (X = H, O, N) in 1 and 4. Spectroscopic information, including vibrational bands and assignments, rotational and centrifugal distortion constants, and vibration–rotation coupling constants, for these molecules was obtained via anharmonic frequency computations at the CCSD/aug-cc-pCVTZ level of theory. Vibrationally averaged ground-state electric dipole moments of 1 and 4 were calculated to be 3.60 and 3.11 D, respectively, which were sufficiently large to allow their experimental and astrochemical characterization by rotational spectroscopy. Thus, 1 and 4 are appropriate candidates for interstellar observation and experimental preparation. The present theoretical information about 1 and 4 and the probable experimental detection of these isomers in the future would significantly improve our understanding of the gas-phase reaction chemistry and astrochemistry of alkaline-earth metals.