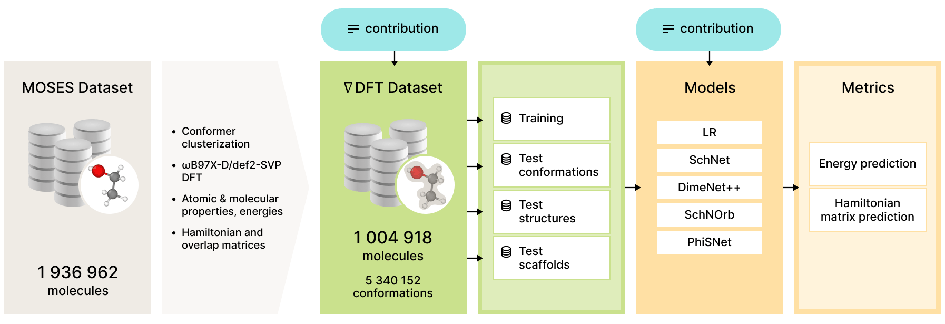
Electronic wave function calculation is a fundamental task of computational quantum chemistry. Knowledge of the wave function parameters allows one to compute physical and chemical properties of molecules and materials. Unfortunately, it is infeasible to compute the wave functions analytically even for simple molecules. Classical quantum chemistry approaches such as the Hartree–Fock method or density functional theory (DFT) allow to compute an approximation of the wave function but are very computationally expensive. One way to lower the computational complexity is to use machine learning models that can provide sufficiently good approximations at a much lower computational cost. In this work we: (1) introduce a new curated large-scale dataset of electron structures of drug-like molecules, (2) establish a novel benchmark for the estimation of molecular properties in the multi-molecule setting, and (3) evaluate a wide range of methods with this benchmark. We show that the accuracy of recently developed machine learning models deteriorates significantly when switching from the single-molecule to the multi-molecule setting. We also show that these models lack generalization over different chemistry classes. In addition, we provide experimental evidence that larger datasets lead to better ML models in the field of quantum chemistry.