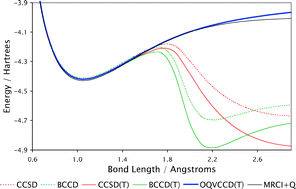
We enhance the recently proposed Optimized-orbital Quasi-Variational Coupled Cluster Doubles (OQVCCD) method for the calculation of ground-state molecular electronic structure by augmenting it with the standard perturbative (T) correction for the effects of connected triple excitations. We demonstrate the OQVCCD(T) ansatz to be outstandingly robust and accurate in the description of the breaking of the triple bond in