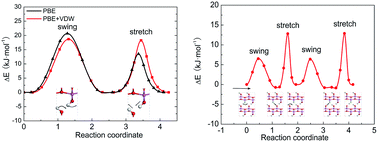
The crystal structure of boehmite (γ-AlOOH) contains a large amount of hydrogen bonds that are joined into chains by sharing hydrogen-bond donor and acceptor oxygen atoms. The hydrogen ions in the hydrogen-bond chains are highly mobile and have complicated structural characterizations, and this feature may well be utilized for proton-conducting applications, but the mechanism is unknown without the dynamic parameters of the hydrogen-transfer processes. We propose probable hydrogen-transfer paths and compute their energy barriers using density functional theory with van der Waals density functionals, on both perfect and vacancy-containing crystal structures. It is revealed that the energy barriers are generally below 21 kJ mol−1 in a perfect crystal, and 14 kJ mol−1 in a vacancy-containing structure. The low energy barriers are indicators of the high proton conductivity of boehmite even at room temperature.